


PrimerHunter User Guide
Version 1.0.1
PrimerHunter is a tool to design highly sensitive and specific primers for virus subtyping via Polymerase
Chain Reaction (PCR). PrimerHunter searches for forward and reverse primers that hybridize to each one of a
given set of DNA sequences called targets (e.g., sequenced isolates from the virus subtype of interest). It
also takes as input a second set of DNA sequences called non-targets (e.g., sequenced isolates from closely
related virus subtypes), and discards primer candidates that hybridize to any of these non-target
sequences. Primer-template hybridization is predicted by performing accurate melting temperature
computations allowing for mismatches. These computations are based on the state-of-the-art
nearest-neighbor model of [5] in conjunction with the fractional programming algorithm
of [2], modified to incorporate salt correction models of [5] and
[3]. Once forward and reverse primers specific to the set of target sequences are selected,
PrimerHunter forms pairs of primers yielding amplification products with length within a user-specified
range. Full details on the algorithm used by PrimerHunter together with
quantitative PCR experiments validating several primer pairs selected using PrimerHunter for
Avian Influenza subtyping are given in [1].
PrimerHunter can be used directly through the web interface provided at
http://dna.engr.uconn. edu/software/PrimerHunter/.
However, for heavier use we recommend downloading the PrimerHunter
source code from the above address and running it locally.
For PrimerHunter questions not addressed in this document,
please contact
Jorge Duitama (jduitama@engr.uconn.edu)
or
Ion Mandoiu (ion@engr.uconn.edu).
To build the executable for PrimerHunter download the source code from
http://dna.engr.uconn.edu/ software/PrimerHunter/PrimerHunter-1.0.0.tar.gz and
execute the following commands
tar -zxvf PrimerHunter-1.0.0.tar.gz
cd PrimerHunter
make
PrimerHunter has been compiled and tested successfully on Gentoo Linux using
the GNU gcc compiler version 3.4.5.
By default PrimerHunter reads target sequences from a fasta file
(specified using the -tf command line parameter) and prints the
results to the standard output. If a fasta file with non target sequences is available, it
should be specified using the -nf command line parameter.
Two sample pairs of target/non-target files are included with the code distribution;
PrimerHunter can be run on these sample files by using the following commands:
./primerhunter -tf sampleTarget.txt -nf sampleNonTarget.txt
respectively
./primerhunter -tf sampleTarget2.txt -nf sampleNonTarget2.txt
PrimerHunter works in two stages. In the first stage the program selects forward and reverse primers
predicted to hybridize strongly with all target sequences and (if available) with none of the non-target
sequences. In addition to enforcing target and non-target hybridization constraints, the first stage of
PrimerHunter also enforces individual primer constraints similar to those used in Primer3 [4],
including constraints on GC Content, 3' end GC Clamp, number of single nucleotide repeats, self
complementarity, and melting temperature against the candidate's perfect complement. In the second stage
PrimerHunter forms pairs of selected primers subject to user-specified constraints on PCR product length
and cross hybridization constraints similar to those implemented in Primer3.
At the end of the first stage, PrimerHunter reports statistics on the number of primer candidates that pass
each design criteria. This allows users to identify criteria that may need to be relaxed to ensure design
success. Since identifying the optimal combination of primer design parameters often requires running
PrimerHunter multiple times, to greatly reduce the design time we recommend first running PrimerHunter with
very relaxed design criteria (in particular, with low minimum melting temperature against targets and high
maximum melting temperature against non targets) and saving detailed candidate primer information into a
file using the -pof command line parameter,1 as in:
./primerhunter -tf sampleTarget2.txt -nf sampleNonTarget2.txt -pof primersSaved.txt
When exploring more stringent primer design criteria, PrimerHunter will run much faster by starting from
the primers info file (specified using the -pf command line parameter as in the example below)
instead of the targets and non-targets files:
./primerhunter -pf primersSaved.txt
The user can instruct PrimerHunter to select primers within a specific regions of input sequences. This
feature is useful when the user has prior knowledge about most discriminative regions between targets and
non-targets, and can also be used to speed-up the primer design process by running parallel processes for
different sequence regions. For cases when target sequences are highly divergent, PrimerHunter allows the
user to relax the constraint that forward and reverse primer candidates must strongly hybridize to all
targets, and require instead that each candidate primer hybridize to a minimum percentage of target
sequences. When PrimerHunter cannot find primer pairs that feasibly amplify all target sequences it
automatically employs a greedy set cover algorithm to select a minimum size set of primer pairs such that
each target sequence is feasibly amplified by at least one primer pair in the set. The non-targets
filtering can be similarly relaxed, allowing selected primers to hybridize to a small percentage of
non-targets. To maintain specificity, primer pairs that feasibly amplify one of the non-target sequences
are discarded before running the greedy set cover algorithm.
PrimerHunter Parameters
Except for the full-stats parameter, each PrimerHunter parameter is specified in the format
-parameterName <
 >. Parameters can be specified in any order on the command line.
>. Parameters can be specified in any order on the command line.
- -tf <
 >
>
Target sequences file in FASTA format (default: none).
-
-nf <
>
Non-target sequences file in FASTA format (default: none).
- -pf <
 >
>
File with candidate primers information from a previous run of PrimerHunter (default: none).
The file has a header line with the number of targets, the number of non targets and the sequence used to
print the positions, all separated by commas. The file then contains three lines for each candidate primer.
The first line
has the following values separated by commas: primer ID, sequence in  to
to  orientation, forward
primer
indicator (
orientation, forward
primer
indicator ( for forward, 0 for reverse) and ID of the target sequence from which the primer comes
from. The second
line has two numbers for each target sequence: the position where the primer is predicted to hybridize
and the primer-target melting temperature (all numbers are separated by commas). The third line contains
the comma separated list of maximum melting temperatures against non target sequences. A sample
candidate primers info file is included with the code distribution and
is also linked to the web interface.
for forward, 0 for reverse) and ID of the target sequence from which the primer comes
from. The second
line has two numbers for each target sequence: the position where the primer is predicted to hybridize
and the primer-target melting temperature (all numbers are separated by commas). The third line contains
the comma separated list of maximum melting temperatures against non target sequences. A sample
candidate primers info file is included with the code distribution and
is also linked to the web interface.
-
-pof <
>
File where the output primers summary information will be stored (default: none).
This option can only be used in command line mode; a primers info file is always generated
when PrimerHunter is executed from the web interface.
-
-minPrimerLength <
 >
>
Minimum length for selected primers (default: 20).
-
-maxPrimerLength <>
Maximum length for selected primers (default: 25).
- -minProdLength <>
Minimum predicted PCR product length for selected primer pairs (default: 75).
This constraint is enforced against all target sequences; product length is computed from
predicted positions for ends of forward and reverse primers.
- -maxProdLength <>
Maximum predicted PCR product length for selected primer pairs (default: 200).
This constraint is enforced against all target sequences; product length is computed from
predicted positions for ends of forward and reverse primers.
- -forwardPrimer <
 >
>
Forward primer sequence suggested by the user (default: none).
When a forward primer is specified, PrimerHunter applies every filter to this
primer, outputs a detailed report about its properties, and does not look
for other forward primers.
- -reversePrimer <
>
Reverse primer sequence suggested by the user (default: none).
When a reverse primer is specified, PrimerHunter applies every filter to this
primer, outputs a detailed report about its properties, and does not look
for other reverse primers.
- -beginPosForward <
 >
>
The start of the range of target sequence positions to be searched for forward primers
(default: 0).
- -endPosForward <>
The end of the range of target sequence positions to be searched for forward primers
(default: 100000). The search ends at the end of target
sequences used to generate candidate primers if the specified position
exceeds their length.
- -beginPosReverse <>
The start of the range of target sequence positions to be searched for reverse primers
(default: 0).
- -endPosReverse <>
The end of the range of target sequence positions to be searched for reverse primers
(default: 100000). The search ends at the end of target
sequences used to generate candidate primers if the specified position
exceeds their length.
- -tmask <
 >
>
A 0-1 mask with 1's indicating, in to order, the candidate primer bases
that must be perfectly matched in predicted primer-target duplexes (default: 11).
The mask is implicitly padded with 0's at the end to match the candidate primer's length.
For example, ``-tmask 1101'' specifies that the first,
second, and fourth -most bases of the primer must be matched exactly.
The mask is used to increase the amplification specificity of selected primers.
- -nmask <
>
A 0-1 mask with 1's indicating, in to order, the candidate primer bases
that must be perfectly matched in predicted primer-non-target duplexes (default: none).
The mask is implicitly padded with 0's at the end to match the candidate primer's length.
For example, ``-nmask 1101'' specifies that the first,
second, and fourth -most bases of the primer must be matched exactly.
The mask is used to speed-up candidate primer filtering based on predicted
hybridizations with non-target sequences.
-
-dmask <
>
A 1-4 mask with 4's indicating, in to order, the positions where
candidate primers have fully degenerate bases (default: 1).
The mask is implicitly padded with 1's at the end to match the candidate primer's length.
The current implementation restricts the number of fully degenerated positions to at
most four. Furthermore, a degenerate base is not allowed at a position required to have a perfect
match in the targets mask.
The degeneracy mask is normally used in conjunction with a complementary
target mask to guarantee a certain number of perfect matches at the end
of selected primers even in the presence of a significant amount of
variability in target sequences.
- -numSourceSeq <
 >
>
Number of target sequences to be used for generating candidate primers (default: 1).
PrimerHunter generates candidate primers from substrings of the first target sequences.
If the specified value is larger than the number of target sequences then all target
sequences are used to generate candidate primers. The default value typically works well,
but increasing the search space for candidate primers may be needed for some
difficult instances.
-
-minCoverageTargets <
 >
>
Minimum percentage of target sequences that must hybridize with each selected primer (default: 100).
Using this parameter automatically activates the set cover algorithm to find the minimum
set of primer pairs collectively covering all target sequences.
- -maxCoverageNonTargets <
>
Maximum percentage of non-targets that can hybridize with a selected primer (default: 0).
This parameter relaxes the non targets filter. In the pairs formation stage,
PrimerHunter discards all pairs for which the two primers are predicted to
simultaneously hybridize to the same non-target sequence.
- -maxSelfScore <>
Maximum score allowed for the local alignment between a primer and its reverse complement
(default: 800). The alignment is performed using the same scoring scheme and code as
in primer3 (a score of  for matches,
for matches, 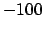 for mismatches, and
for mismatches, and  for gaps).
for gaps).
- -maxEndScore <>
Maximum score allowed for the -anchored global alignment of a
primer and its reverse complement (default: 300). The alignment is
performed using the same scoring scheme and code as in primer3.
-
-minGCContent <
>
Minimum percentage of GC content allowed for a primer (default: 0).
-
-maxGCContent <
>
Minimum percentage of GC content allowed for a primer (default: 100).
- -gcClamp <>
Number of required consecutive GC bases in the end of each primer (default: 0).
- -maxPolyX <>
The maximum allowed length of a mononucleotide repeat in a primer (default: 5).
For example, the sequence AAAAAA can not appear in a primer under the default settings.
- -primersConc <
 >
>
Primer concentration (M) to be used in melting temperature computations (default: 0.0000008).
- -templateConc <
>
DNA template concentration (M) to be used in melting temperature computations (default: 0).
-
-saltConc <
>
Salt concentration (M) to be used in melting temperature computations (default: 1).
-
-saltCorrMethod <>
Salt correction method: 1 for Santalucia's formula, 2 for Owczarzy's formula (default 1).
- -minTempTargets <>
Minimum melting temperature (in degrees Celsius) for primer-target hybridizations (default: 40).
- -maxTempTargets <>
Maximum melting temperature (in degrees Celsius) for primer-target hybridizations (default: 70).
- -maxTempNonTargets <>
Maximum melting temperature (in degrees Celsius) for primer-non-target hybridizations
at locations where the primer matches the non-target sequence according to the
non-target mask (default: 50).
-
-deltaTempNonTargets <>
Minimum difference (in degrees Celsius) between the highest primer melting
temperature against a non-target sequence
and the lowest primer melting temperature against a target sequence (default: 0).
- -maxPairTempDiff <>
Maximum difference (in degrees Celsius) between the melting temperatures of the two primers
in a pair against each target sequence (default: 40).
- -primersLabel <
 >
>
Label to be used in forming primer names (default: ``P'').
The algorithm generates the complete name for each primer from the string
``Forward'' or ``Reverse'', depending of the type of primer, followed by a _ character,
followed by the string specified using -primersLabel, followed
by another _ character, and finally a consecutively generated integer.
Using the default settings, a complete primer name will look like ``
 ''.
''.
-
-primersLabel
If this flag is included, the program will not throw a candidate primer until every test is performed.
The number of primer candidates passing each test can then be used to identify overly
restringent filtering criteria.
- The web interface can optionally receive a valid e-mail address at which a notification will be sent when
PrimerHunter finishes its computation. This option is not available
in the command line mode.
- Web interface modified to make optional the e-mail address. An answer page was built to display the results as soon as the process finishes.
- Initial release
- 1
-
J. Duitama, D.M. Kumar, E. Hemphill, M. Kahn, I.I. Mandoiu, and C.E. Nelson.
PrimerHunter: A primer design tool for PCR-based virus subtype
identification.
Nucleic Acids Research, submitted, 2008.
- 2
-
M. Leber, L. Kaderali, A. Schonhuth, and R. Schrader.
A fractional programming approach to efficient DNA melting
temperature calculation.
Bioinformatics, 21(10):2375-2382, 2005.
- 3
-
R. Owczarzy, Y. You, B.G. Moreira, J.A. Manthey, L. Huang, M.A. Behlke, and
J.A. Walder.
Effects of sodium ions on DNA duplex oligomers: Improved
predictions of melting temperatures.
Biochemistry, 43(12):3537-3554, 2004.
- 4
-
S. Rozen and H.J. Skaletsky.
Primer3 on the WWW for general users and for biologist programmers.
In S. Krawetz and S. Misener, editors, Bioinformatics Methods
and Protocols: Methods in Molecular Biology, pages 365-386. Humana Press,
Totowa, NJ, 2000.
- 5
-
J. SantaLucia and D. Hicks.
The thermodynamics of DNA strucutural motifs.
Annual Review of Biophysics and Biomolecular Structure,
33:415-440, 2004.
2008-11-10