


Next: Local Structural Alignment algorithm
Up: Structural Alignment algorithm based
Previous: Structural Alignment algorithm based
Description of protein structure in PDB format
The PDB file for a protein structure is a text description of the 3D-coordinates
of the atoms/residues with the protein is comprised of. The file consists of
a linear list 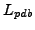 list of atoms which are part of the protein and corresponding
3-D coordinates of each atom, these atoms are part of the residues which correspond to
the 20 Amino-acids.
list of atoms which are part of the protein and corresponding
3-D coordinates of each atom, these atoms are part of the residues which correspond to
the 20 Amino-acids.
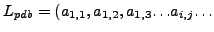 ), and
), and 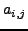 is the
is the 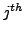 atom in residue
atom in residue 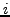 , a very important fact to be noticed at this point is
that the list
is partially ordered with respect to the residue numbers,
, a very important fact to be noticed at this point is
that the list
is partially ordered with respect to the residue numbers,
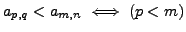 , although there is an ordering in the residues the
atoms within each of these residues may not follow any order. Thus given two
instances of a same protein PDB structure
, although there is an ordering in the residues the
atoms within each of these residues may not follow any order. Thus given two
instances of a same protein PDB structure
 and
and
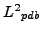 , the ordering
of the atoms within each residue(residue's will be in same order) may be different, as an
example the atoms in
, the ordering
of the atoms within each residue(residue's will be in same order) may be different, as an
example the atoms in 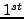 residue of
may be ordered as
residue of
may be ordered as
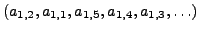 but the atoms in the same residue of
can be ordered as
but the atoms in the same residue of
can be ordered as
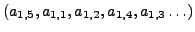 . The
reason for this variation is mainly due to different frames of reference during X-ray
crystallography. The variation of the ordering of the atoms within the same residue makes structural alignment
algorithms which consider the sidechain conformations non-trivial. In the next few sections
we will see how our algorithm overcomes this ordering issue when side-chain conformations are
considered.
. The
reason for this variation is mainly due to different frames of reference during X-ray
crystallography. The variation of the ordering of the atoms within the same residue makes structural alignment
algorithms which consider the sidechain conformations non-trivial. In the next few sections
we will see how our algorithm overcomes this ordering issue when side-chain conformations are
considered.
Next: Local Structural Alignment algorithm
Up: Structural Alignment algorithm based
Previous: Structural Alignment algorithm based
Vamsi Kundeti
2007-10-10